CS
414
Team
17
Friday
January 26, 2000
![]()
Software
Specification
![]()
Protein Sequence Analyzer
Modifications
Don Metzler
Kathy
Repine
Chris
Schornstein
![]()
1.
Introduction
1.1 Purpose
The purpose of this document is to describe the
requirements for the modifications to be made to the Protein Sequence Analyzer,
the data model will be used, the process to be followed, and the remaining
tasks that need to be completed.
1.2 Project Description
1.2.1 Description
The system, which
runs in a UNIX environment, consists of four programs that are written in C and
a bash shell script that links the four programs together. The programs analyze various proteins in an
attempt to learn about and understand the similarities between them, and thus
between organisms that produced those proteins. The proteins are treated as vectors in a high-dimensional
space. Similar proteins point in
similar directions, therefore the cosine of the angle between two proteins can
be used to measure their likeness.
Software exists (the Phylip package neighbor program) that takes these
similarities as a distance matrix and converts them into a tree.
When a user does
analysis with the current system they must tune several parameters by hand, which
can make the entire task very tedious.
The first parameter the user can change is the type of peptide sequences
to analyze. The current options are
tripeptides or tetrapeptides, where a peptide is a protein sequence. However, there is a desire to be able to do
both tri and tetrapeptides during the same analysis. This is a task that this project aims to allow for by way of
allowing multiple peptide sizes to be analyzed at the same time.
The next parameter
the user has to modify is the number of dimensions to use when performing a
singular value decomposition (SVD) on the peptide frequency matrix. The SVD is used to get only the relevant
information from the high dimension peptide frequency, which simplifies
analysis. The peptide frequency matrix is
a matrix that contains the frequencies of each of the peptides present in the
test data. The manner in which the
current program handles this is it will perform the SVD and run through the
rest of the analysis and produce a gene or protein tree depending user input.
This tree will either be correct and contain all related proteins on closely
related branches, or it will be incorrect and have different proteins
interspersed amongst each other. In this case the user must change the number
of dimensions and perform the SVD and resulting analysis again. This entire
process can be very time consuming. This project plans on implementing a
feedback loop that will automate the entire process of picking the correct
number of dimensions for the SVD calculation until the resulting analysis
produces a correct tree.
1.2.2 Scope
The
majority of the project will be modifications to existing code, with some minor
additions to functionality.
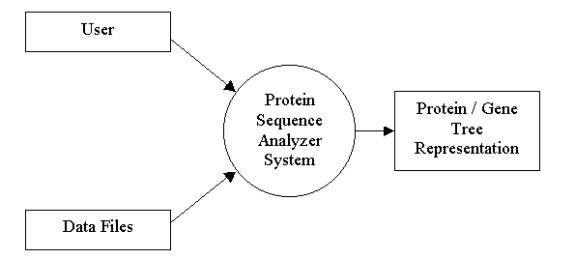
Figure 1.2.2a Context Diagram for the Protein Sequence Analyzer Program
2.
Data Model
2.1 Considerations
The goal of the protein
sequence analyzer system is to analyze a set of protein data and produce a
graphical representation. Depending on
user input, it can be represented in the form of either a protein tree or a
gene tree. Therefore, the main input of
the system is a single file that contains all of the protein data that will be
used in the analysis. This data is then
sent through a number of linked programs (see figure 3.2a) that perform the
analysis and ultimately produce a file containing a tree in New Hampshire
format, a data format for storing trees.
These are then easily converted to a graphical tree. Although this is not part of our project,
many software packages exist that will satisfactorily do this conversion.
2.2 Diagram
The diagram below depicts the entity relationship diagram of the protein sequence analyzer system. It shows the relationships between the main entities in the system and how they relate to each other.
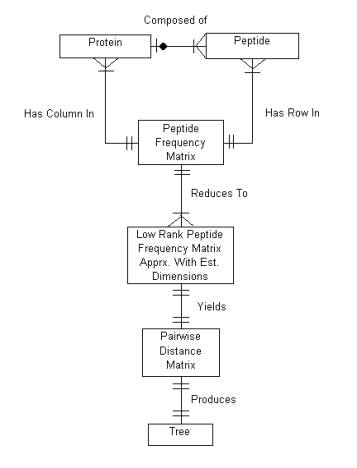
Figure 2.2a Entity Relationship Diagram for the Protein Sequence Analyzer Program
2.3 Requirements
The features of the
protein sequence analyzer system that are to be added will not require any
significant modifications to the existing data flow path. Several small modifications to the script
that links the programs together may be necessary. With the addition of the shift program, tree rating, and course
of action analysis program, changes need to be made in order to ensure the
inputs to these programs are in the correct format. These changes, however, will be minimal since the inputs to the
new modules will be available from previous steps in the script. Nothing else in way of data flow or data
storage will need to be changed; the same input file type and format will still
be valid after the new features are implemented. The only foreseeable changes
for the data model would be larger data sets, but as long as the input file
keeps its current format its size will not affect the general performance of
the system.
3.
Process Model
3.1. Considerations
The project consists of several module tied together with a script. Most of the modules already exist and don’t require any modification. There are only three completely new modules: the shift program, tree rating, and course of action analysis. “aa code” is the only existing module that will require any modifications. The script that links all the modules together and controls the flow of information will need to be updated to include the new modules and to handle the feedback loop from course of action analysis to shift.
3.2 Diagram
The data flow diagram below shows each of the
modules and what data flows between them.
The diagram explains the input format each module expects and describes
the format of the data each module outputs, thereby showing how each module
modifies the data and how each module is related to the others.
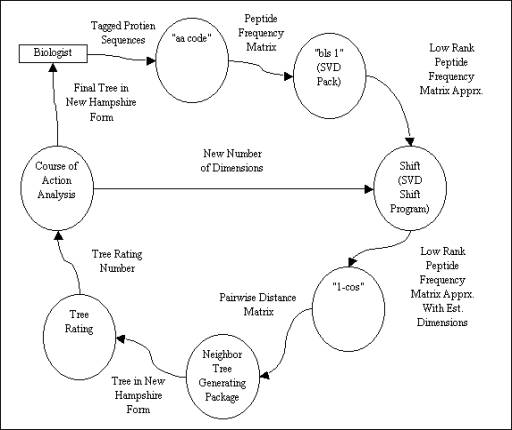
Figure 3.2a Data Flow Diagram for the Protein Sequence Analyzer Program
3.3 Requirements
The three new modules
and the script enhancements are the bulk of the project. The shift module should be publicly
available software that fits well into the script. The tree rating module should take a tree in New Hampshire form
and rate it based on how distinctly sets of tagged proteins are separated into
families. The recommended way of
accomplishing this is to compact the tree into a string of symbols, where each
symbol represents a specific tag.
Ideally, there would only be one symbol for each tag, thus representing
that each of the identically tagged proteins appear in the same area of the
tree. However, this might not always
occur. Therefore, the rating should be
based on how interspersed the different tags are. The course of action analysis should take the tree rating and
decide if the tree is as good as it will get or if changing the number of
dimensions will improve it. If course
of action analysis decides to change the number of dimensions, it needs to decide
whether to increase or decrease them and by how much. These directions and numbers will be determined empirically and
should be easy to change or update.
4.
Revised Plan
4.1 Revisions
There are several additions to the program that are possible. Two of these features are implementing a graphical user interface, and the ability to include entropy calculations in the analysis of the data sets. Although these additions are possible, they are not within the scope of this project and therefore will not be implemented.
4.2 Tasks List
These are the tasks that must be completed:
· Update Old Code: The old in-house code uses a lot of hacks and lacks comments, thus it must be updated.
· Script: The script must be modified to include the added functionality of the additional modules and the new feedback loop.
· Shift Program: A publicly available SVD shift program, that can easily integrated into the script, must be found.
· Tree Evaluation: The tree must be rated based on how well it separates tagged proteins into distinct groups. From that rating, course of action analysis must decide which dimension to feedback to the shift program, if any. The empirical numbers for course of action analysis must be calculated.
· Multi-Sized Protein Sequences: The “aa code” module must be modified to handle peptide sequences of varying size simultaneously.
· Feedback Loop: The separate parts of the feedback loop mentioned above must be integrated together.
·
Testing:
Verifying the feedback loop yields the best tree
4.3 Remaining Phases
The breakdown of the tasks into project phases is as follows:
· Phase 2: Update old code, script modifications, begin tree evaluation, and prototype #2
· Phase 3: N-grams, tree evaluation (continued), shift program, feedback loop
· Phase 4: Testing, and delivery of final product
4.4 Schedule
|
Phase |
Estimated Time
for |
|
Phase 2 |
3 weeks |
|
Phase 3 |
5 weeks |
|
Phase 4 |
5 weeks |
5.
Conclusion
5.1 Summary
The improved protein sequence analyzer program will
include the existing program along with several new modules. One of these new
modules will implement a feedback loop in which the user will not have to
adjust the number of dimensions of the frequency matrix by hand, as is the
current situation. Also, the user of the system will be able to run analysis on
different size peptide sequences, thus expanding the usefulness of the program
as a whole. The paper laid out how these modules will be added to the existing
program, the data model involved, the process to be followed, and the remaining
tasks to be completed.
5.2 Conclusion and Recommendation
Given the current schedule and the improvements that
were proposed in this document, it is concluded that the project can be
finished within the given timeframe. Since the added functionality will greatly
enhance the program it is recommended that that the project continue upon the
current schedule until completion.
5.3 Contacts
Any additional questions can be directed at:
Don Metzler (Donald.A.Metzler@Rose-Hulman.edu)
Kathleen Repine (Kathleen.M.Repine@Rose-Hulman.edu)
Chris Schornstein (Christopher.J.Schornstein@Rose-Hulman.edu)
Gary Stuart, the client at Indiana State University, can be contacted at (812) 237-7898 or lsstuar@scifac.indstate.edu.